Search

A $350,000 Cure4 Cystic Fibrosis grant is set to propel the Wal-yan Respiratory Research Centre’s Phage WA program forward, supercharging its fight against antimicrobial resistant (AMR) lung infections in people with Cystic Fibrosis (CF) using cutting-edge phage therapy.

A promising new treatment pioneered in Western Australia for people with cystic fibrosis has commenced testing in a clinical trial in the United States and Australia.
Promising results from an Australian-led clinical trial could drastically change the way we care for young children with cystic fibrosis (CF).
The Kids Research Institute Australia spin-off company, Respirion, received $20 million in funding to develop a promising new therapy.

The family of two girls with cystic fibrosis are hopeful after The Kids Research Institute Australia spin-off company, Respirion, receives $20 million in funding to develop a promising new therapy.

A The Kids Research Institute Australia spin-off company has received $20 million from the Medical Research Commercialisation Fund to develop a promising new therapy for the treatment of Cystic Fibrosis.

The Kids researchers are pioneering an exciting new approach to clinical trials, which aims to fast-track the best treatments for people with rare and complex diseases.
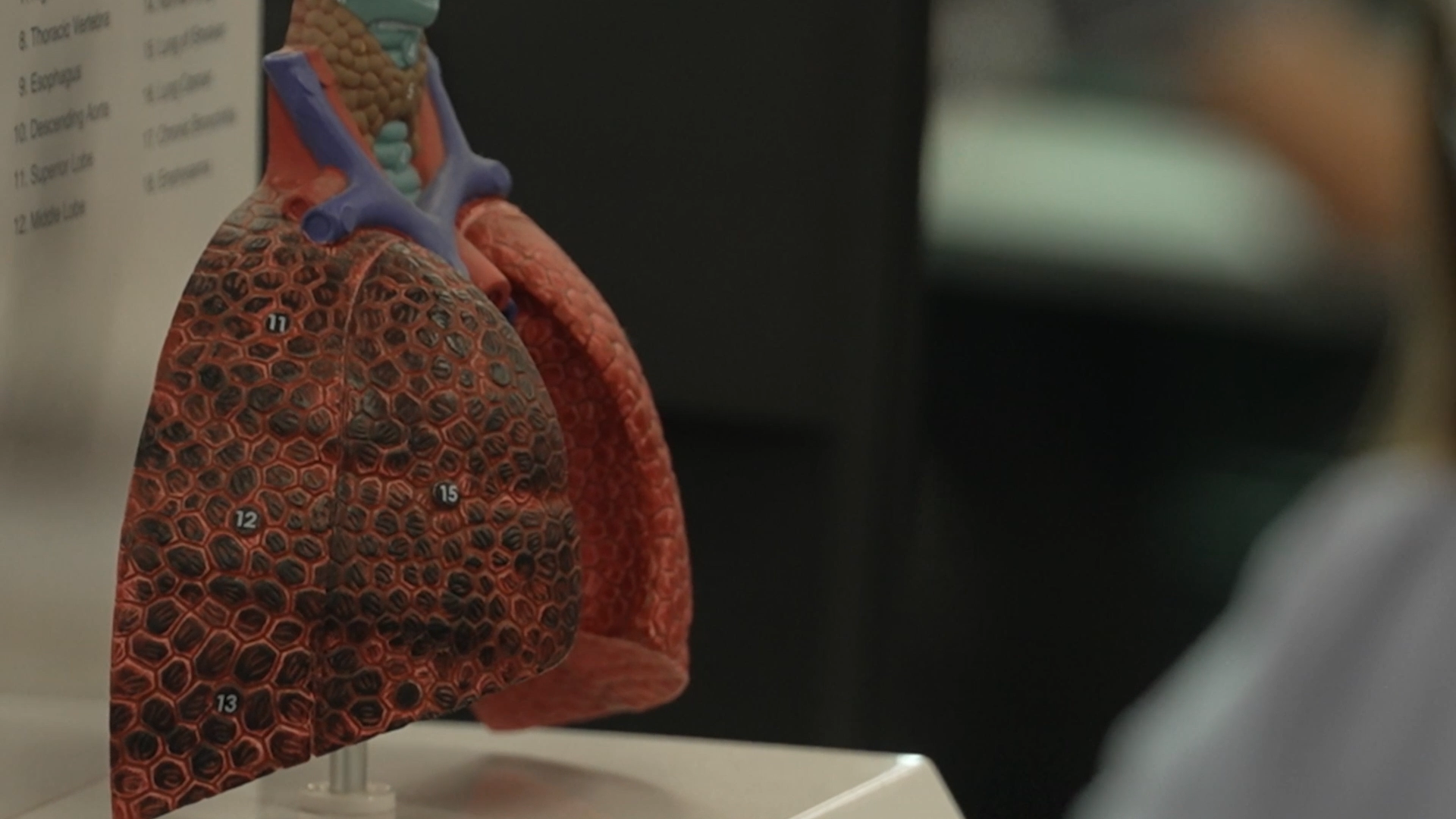
A population of neutrophils recruited into cystic fibrosis (CF) airways is associated with proteolytic lung damage, exhibiting high expression of primary granule exocytosis marker CD63 and reduced phagocytic receptor CD16. Causative factors for this population are unknown, limiting intervention. Here we present a laboratory model to characterize responses of differentiated airway epithelium and neutrophils following respiratory infection.
Infants with cystic fibrosis (CF) develop structural lung disease early in life, and viral infections are associated with progressive lung disease. We hypothesized that the presence of respiratory viruses would be associated with structural lung disease on computed tomography (CT) of the chest in infants with CF.
Primary ciliary dyskinesia (PCD) is a rare, progressive, inherited ciliopathic disorder, which is incurable and frequently complicated by the development of bronchiectasis. There are few randomised controlled trials (RCTs) involving children and adults with PCD and thus evidence of efficacy for interventions are usually extrapolated from people with cystic fibrosis.